You are here: Urology Textbook > Kidneys > Autosomal dominant polycystic kidney disease
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
Definition of Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) is a common monogenic multisystem disorder characterized by progressive development of multiple renal cysts, progressive decline in kidney function during adulthood, hepatic cysts, and intracranial aneurysms.
Epidemiology
- Incidence 1:2000
- 7–15% of dialysis patients are suffering from ADPKD
Etiology of Autosomal Dominant Polycystic Kidney Disease
Genetics:
Most patients have pathogenic variants in two genes: in 85–90%, PKD1 (chromosome 16, encoding polycystin-1) is affected, and in 10–15%, PKD2 (chromosome 4, encoding polycystin-2) is affected. PKD2 mutations typically cause a milder disease course, with later onset and slower progression. More rarely, other genetic alterations cause ADPKD or an ADPKD-like phenotype. Autosomal dominant inheritance with very high penetrance is typical, so that 50% of the children of affected patients inherit the disease. The disease develops according to Knudson’s two-hit theory: one affected allel is inherited, and the second allel is altered later by a spontaneous mutation, which explains the long symptom-free latency before disease onset.
Pathophysiology:
Polycystin-1 and polycystin-2 have essential functions in signal transduction and in forming the primary cilium of the tubular epithelial cells. Defective polycystin leads to the tubular epithelium's proliferation and cyst formation; every part of the nephron may be affected. Similar mechanisms may damage blood vessels and other organ systems. The disease leads to the activation of mTOR signaling, enhances cAMP production, and increases ion and fluid secretion into the renal cystic lumen. The vasopressin V2-receptor blocker tolvaptan inhibits ADH dependent cAMP production, slows the increase in kidney volume, and delays the development of chronic kidney disease.
Progression factors:
The normal (bilateral) kidney volume is usually below 400 ml. In ADPKD, increasing renal volume is associated with decreasing renal function. The kidney volume can be measured either by MRI or renal ultrasound imaging: the greater the kidney volume, the worse the prognosis for renal function. The time to end-stage renal disease for a 30-year-old man depends on the renal volume: 10 years (renal volume 2000 ml), 18 years (1500 ml) or 21 years (1000 ml) (Kuehn et al., 2015).
Pathology:
Autosomal dominant polycystic kidney disease leads to enlarged kidneys with multiple cysts [fig. polycystic kidneys]. The cysts are a few millimeters to a centimeter in size and derive from tubules of the nephron. The epithelium of the cyst corresponds to its origin. Cysts also develop in other organ systems; see section signs and symptoms [fig. liver cysts].
Signs and Symptoms
Onset of disease:
Symptoms start at the age of 30 to 50 years. Renal ultrasound and genetic screening lower the average age of initial diagnosis due to the discovery of the asymptomatic disease.
Symptoms:
- Arterial hypertension
- Hematuria
- Proteinuria
- Flank pain and fever (cyst rupture or cyst infection)
- Nephrolithiasis
- Gastrointestinal symptoms due to large kidneys
Manifestations in other organs:
- Cysts in the liver, pancreas, and other organs
- Aneurysms of the cerebral arteries with a risko of subarachnoidal hemorrhage.
- Colonic diverticula
- Mitral valve prolapse, thoracic aortic ectasia.
Diagnostic Workup of Autosomal Dominant Polycystic Kidney Disease
Family history:
Covering at least three generations.
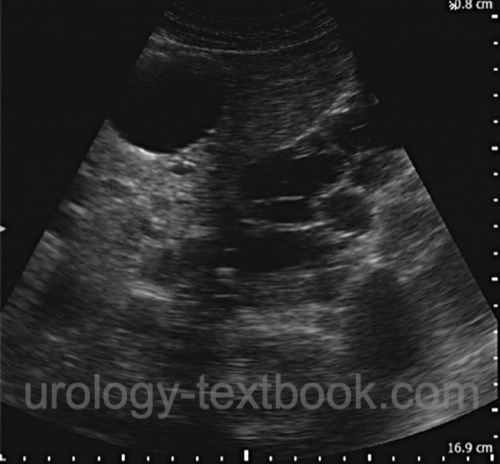
Ultrasonography:
In adults with a positive family history, abdominal ultrasonography is the recommended first-line examination. The kidneys, liver, pancreas, and spleen should be assessed for organ cysts. Determination of bilateral kidney volume is important for prognostic assessment regarding the later development of kidney failure in patients with manifest ADPKD. Age-dependent criteria improve diagnostic accuracy: in patients 15–39 years of age, at least 3 renal cysts in total are consistent with ADPKD; in patients 40–59 years of age, at least 2 cysts in each kidney; and in patients 60 years or older, at least 4 cysts in each kidney. To exclude ADPKD in individuals with a positive family history, the following applies: at 15–39 years of age, the absence of cysts or the presence of no more than 1 cyst largely excludes the disease; at 40–59 years of age, the presence of no more than 2 cysts in total largely excludes the disease.
 |
Laboratory examinations:
- Urine analysis for proteinuria
- Creatinine or/and cystatin C to evaluate renal function.
- Genetic evaluation: for the differential diagnosis of unclear cystic kidney disease and as a predictive diagnosis in case of a positive family history and atypical imaging.
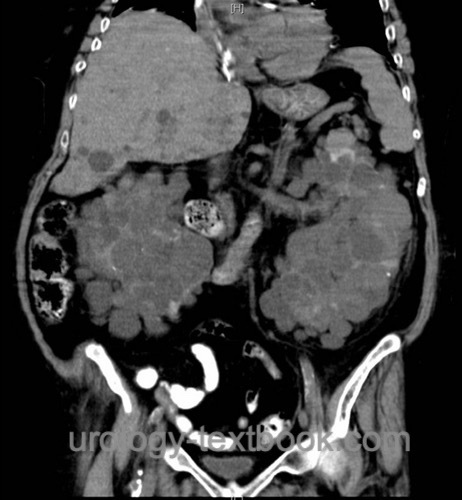
CT or MRI:
- Abdominal imaging: in patients with flank pain or suspect ultrasound imaging.
- Cranial imaging: to estimate the risk of intracranial bleeding due to aneurysms.
 |
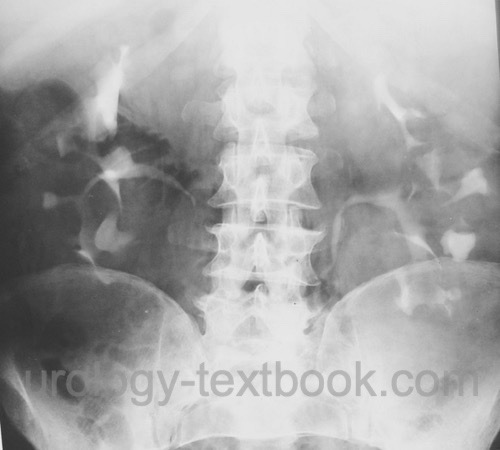
Intravenous urography:
Intravenous urography is only indicated, if abdominal CT is not available. Urography reveals bilateral renal enlargement, calyceal stretching from the cysts, and Swiss cheese aspect in the nephrographic phase.
 |
Treatment of Autosomal Dominant Polycystic Kidney Disease
Medical Treatment:
Early therapy of arterial hypertension with ACE inhibitors or angiotensin-II receptor antagonists. Low-salt diet. Protein reduction does not improve the prognosis. The vasopressin (V2) receptor antagonist tolvaptan has been approved in patients at high risk of progression to slow down disease progression (Torres et al., 2012). Tolvaptan delays the onset of dialysis by approximately one year for every five years of treatment. Polyuria (sometimes over 7 l/d) and idiosyncratic hepatotoxicity are problematic side effects of tolvaptan that require intensive patient monitoring.
Management of Flank Pain:
Nephrolithiasis, bleeding, or infection of cysts may be responsible for flank pain. In cyst infection, prolonged treatment with lipophilic antibiotics for several weeks is necessary. If imaging can reveal altered cysts, percutaneous management (cyst aspiration and sclerotherapy) or laparoscopic deroofing of cysts may be helpful.
Treatment of Renal Failure:
Hemodialysis and kidney transplantation are the treatment alternatives. Nephrectomy is often necessary before renal transplantation: either to create room for the transplant or if recurrent symptoms from the enlarged polycystic kidneys are present.
Experimental Treatment:
Pharmacologic inhibition of tubular epithelial cell proliferation by signal transduction inhibitors is under investigation. Previous studies with mTOR inhibitors such as everolimus or tyrosine kinase inhibitors have not demonstrated a protective effect on kidney function.
Prognosis of Autosomal Dominant Polycystic Kidney Disease (ADPKD):
The prognosis has improved due to better treatment options.
Renal failure:
The median age for end-stage renal disease is in the fifth decade (PDK1 mutation) or seventh decade (PDK2 mutation).
Cerebral hemorrhage:
9% of patients with ADPKD die of subarachnoid hemorrhage (ruptured brain aneurysm). In addition, cerebral hemorrhage due to malignant hypertension is possible.
| ARPKD | Index | Kidney diseases |
Index: 1–9 A B C D E F G H I J K L M N O P Q R S T U V W X Y Z
References
Devuyst O, Torres VE, u. a. KDIGO 2025 Clinical Practice Guideline for the Evaluation, Management, and Treatment of Autosomal Dominant Polycystic Kidney Disease (ADPKD). Kidney International. 2025.
 Deutsche Version: ADPKD
Deutsche Version: ADPKD
Urology-Textbook.com – Choose the Ad-Free, Professional Resource
This website is designed for physicians and medical professionals. It presents diseases of the genital organs through detailed text and images. Some content may not be suitable for children or sensitive readers. Many illustrations are available exclusively to Steady members. Are you a physician and interested in supporting this project? Join Steady to unlock full access to all images and enjoy an ad-free experience. Try it free for 7 days—no obligation.
New release: The first edition of the Urology Textbook as an e-book—ideal for offline reading and quick reference. With over 1300 pages and hundreds of illustrations, it’s the perfect companion for residents and medical students. After your 7-day trial has ended, you will receive a download link for your exclusive e-book.