You are here: Urology Textbook > Testes > Disorders of sex development > Congenital adrenal hyperplasia
Congenital Adrenal Hyperplasia: Diagnosis and Treatment
Definitions
Congenital adrenal hyperplasia (CAH) is an autosomal recessive inherited enzyme defect of steroid biosynthesis, which leads to excessive ACTH elevation due to cortisol deficiency (Merke et al., 2005). Adrenal steroids proximal to the enzyme defect are elevated (see also figure steroid biosynthesis) and lead to adrenal hyperplasia. Symptoms differ on the underlying enzyme defect and the sex of the patient.
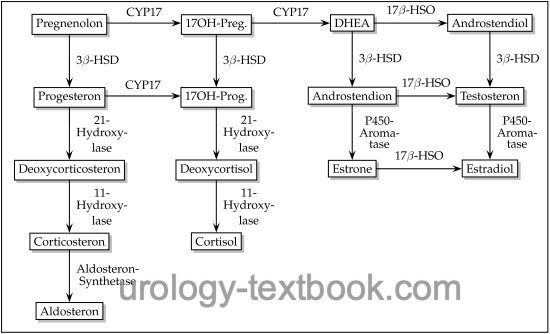 |
Etiology, Signs, and Symptoms of Congenital Adrenal Hyperplasia
21-Hydroxylase Deficiency:
21-hydroxylase deficiency is the most common enzyme defect (95%), with an incidence of 1:14.000 births. Two clinical courses are possible depending on the severity of the enzyme defect: virilization or virilization with salt wasting due to decreased aldosterone synthesis.
Symptoms of 21-hydroxylase deficiency without salt loss:
Girls present with mild virilization (normal female genitals at birth with later clitoral enlargement and hirsutism) or severe virilization (masculinization of the genitals at birth). Boys are born without symptoms, later rapid growth, early puberty, small testes, and early growth arrest.
Symptoms of 21-hydroxylase deficiency with salt loss:
Neonatal emergency: loss of fluid and salt from the 5th day of life due to the prevented aldosterone and cortisone synthesis. Hyponatremia and hyperkalemia are typical, furthermore acidosis, dehydration, masculinization in female newborns, large genitalia in male newborns. Prompt diagnosis is critical to ensure adequate treatment.
Diagnosis of 21-hydroxylase deficiency:
Elevated 17-hydroxyprogesterone, androgens (androstenedione, DHEA, and testosterone), and ACTH. Low serum concentration of cortisone. In patients with salt wasting, aldosterone is decreased despite increased renin activity.
11β-Hydroxylase Deficiency:
11β-hydroxylase deficiency causes 5% of congenital adrenal hyperplasia. The enzyme defect prevents cortisol synthesis and produces excessive 11-deoxycorticosterone and 11-deoxycortisol. Hypertension is common, and severe enzyme deficiency increases adrenal androgens with virilization.
Rare enzyme defects:
Deficiency of 3β-hydroxysteroid dehydrogenase, 17α-hydroxylase, and steroidogenic acute regulatory protein (StAR) also leads to impaired testosterone biosynthesis:
StAR deficiency:
Mutations in the steroidogenic acute regulatory protein (StAR) lead to an impairded cholesterol transfer within the mitochondria and result in mineralcorticoid, glucocorticoid and sex steroid deficiency. Large lipid-laden adrenals displace the kidneys caudally. Male patients develop female external genitalia with a blind-ending vagina without uterus. Female patients present with normal genital organs. Marked (potentially lethal) adrenal insufficiency develops within weeks after birth in both sexes. Early diagnosis and hormone substitution are essential.
3β-hydroxysteroid dehydrogenase deficiency:
3β-hydroxysteroid dehydrogenase deficiency leads to the lack of conversion of pregnenolone into progesterone, DHEA into androstenedione, and androstenediol into testosterone. There are different manifestations of the enzyme defect. In the classic (pronounced) form, the deficiency of aldosterone, cortisol, testosterone, and estradiol causes salt loss, virilization of female patients, and lack of virilization of male patients. Milder forms do not cause salt loss, and the symptoms are nonspecific (including hypospadias and micropenis in boys and hirsutism and infertility in girls).
17α-hydroxylase deficiency:
17α-hydroxylase deficiency leads to impaired synthesis of 17-hydroxyprogesterone and 17-hydroxypregnenolone, resulting in a deficiency of cortisol and sex steroids. Mineral corticoids (deoxycorticosterone and corticosterone) are greatly increased, causing hypertension, hypokalemia, and alkalosis. Male patients have female or ambiguous external sex characteristics. Female patients present with normal genitalia; later, ovarian failure and infertility develop.
Testicular adrenal rest tumor (TART):
Ectopic remnants of adrenal tissue in the testes proliferate under the influence of increased ACTH, especially in patients with insufficient medical therapy. Infertility (azoospermia is possible) and a palpable benign testicular tumor develop. Palpation and ultrasonography cannot distinguish TART from germ cell tumors. A supranormal substitution with cortisone leads to a pronounced regression of the tumors. The bilateral findings and the response to cortisone are decisive, surgery is unnecessary.
Treatment of Congenital Adrenal Hyperplasia
Acute therapy for Addison's crisis with salt loss includes administration of cortisone, fluid and electrolytes. Lifelong substitution of cortisone and fludrocortisone is necessary. The dosage depends on hormone concentrations, electrolytes, blood pressure, the patient's well-being and growth, and signs of virilization. In female patients with marked masculinization, early corrective surgery such as clitoral reduction may be performed, but this is controversial if the child is unable to give consent. Correct hormone adjustment is essential for the fertility of affected men.
| Ovotesticular DSD | Index | Androgen insensitivity syndromes |
Index: 1–9 A B C D E F G H I J K L M N O P Q R S T U V W X Y Z
References
C. Radmayr, G. Bogaert, H. S. Dogan, and Tekgü, “EAU Guidelines: Paediatric Urology,” 2022. [Online]. Available: https://uroweb.org/guidelines/paediatric-urology/.
 Deutsche Version: Enzymdefekte des adrenogenitalen Syndroms
Deutsche Version: Enzymdefekte des adrenogenitalen Syndroms
Urology-Textbook.com – Choose the Ad-Free, Professional Resource
This website is designed for physicians and medical professionals. It presents diseases of the genital organs through detailed text and images. Some content may not be suitable for children or sensitive readers. Many illustrations are available exclusively to Steady members. Are you a physician and interested in supporting this project? Join Steady to unlock full access to all images and enjoy an ad-free experience. Try it free for 7 days—no obligation.
New release: The first edition of the Urology Textbook as an e-book—ideal for offline reading and quick reference. With over 1300 pages and hundreds of illustrations, it’s the perfect companion for residents and medical students. After your 7-day trial has ended, you will receive a download link for your exclusive e-book.